
Streamlining Antibody Discovery: From NGS to Functional Validation
High-Throughput In Vitro Antibody Discovery
Introduction
The primary objective of high-throughput antibody discovery is the delivery of a diversified panel of developable lead candidates. Traditional workflows often fail to meet this objective due to a critical analysis gap—the disconnect between massive NGS datasets and the limited capacity of downstream characterization. Here, we outline a scientifically validated workflow utilizing the Platforma ecosystem. By bridging upstream antibody annotation with downstream functional prediction, this protocol mitigates the risk of selecting redundant or liable scaffolds, ensuring the final candidate pool is maximized for both epitope coverage and developability.
Why NGS for antibody discovery?
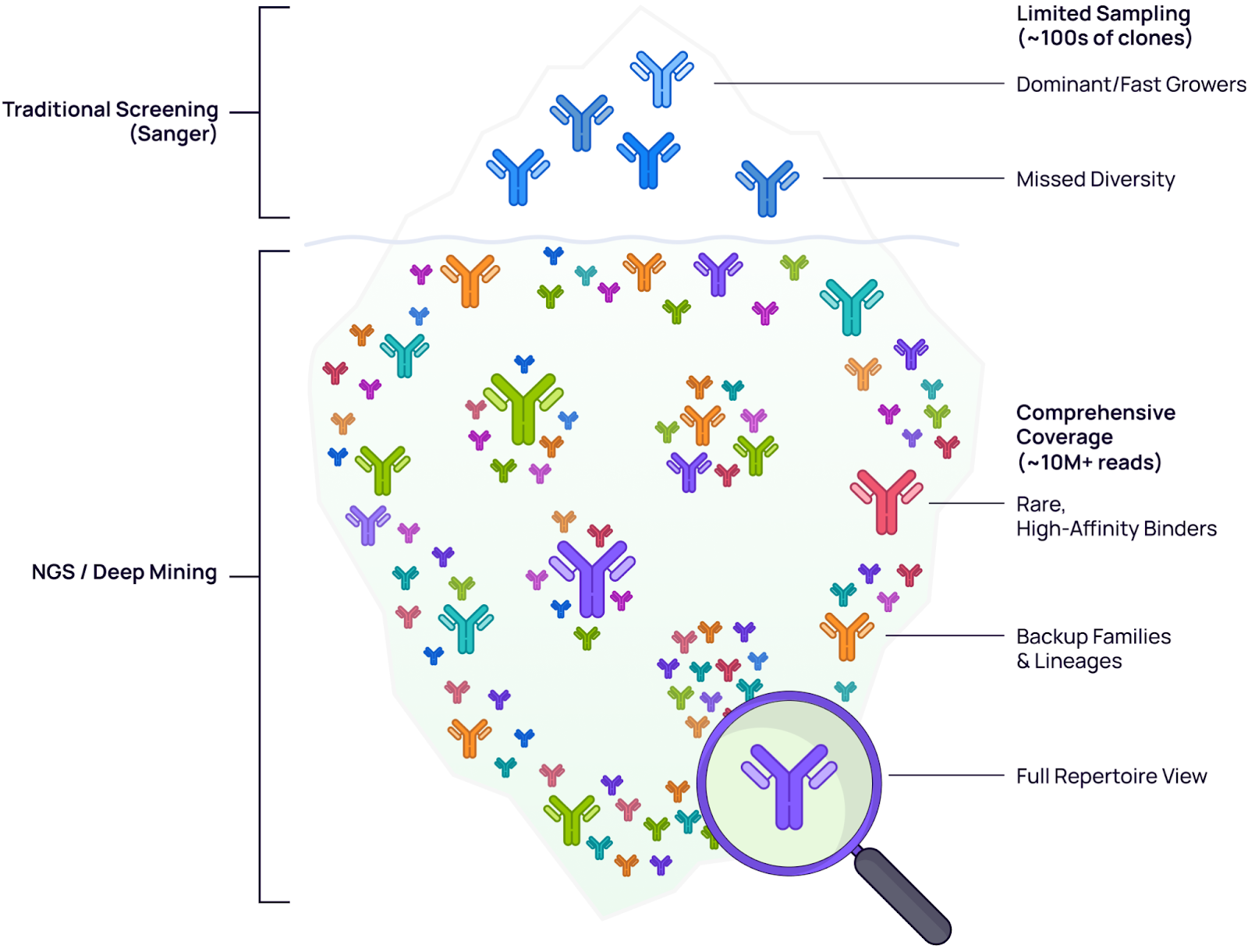
Traditional screening methods typically sample only 100–300 clones—capturing just the tip of the iceberg. This limited sampling introduces significant bias toward fast growers—clones that replicate efficiently in the host system but may lack superior binding affinity. Deep mining (~10M+ reads) with NGS provides the sequencing depth and coverage needed to access the submerged diversity, revealing rare high-affinity binders and distinct backup/alternative lineages that are invisible to shallow screening methods.
Methodology
This workflow enables the end-to-end discovery of VHH, scFv, and Fab candidates within a managed computational environment. Prioritizing algorithmic transparency over black-box solutions, the system is built upon the Platforma SDK and utilizes the proprietary P-frame architecture (columnar and bioinformatics-native storage layer) to ensure data integrity and traceability.
The underlying logic is transparent and open source [1]. At its core, the pipeline performs the sequence of steps for large-scale antibody discovery: annotation, clustering, enrichment analysis and developability assessment. On top of this established workflow, Platforma introduces two innovations that significantly enhance discovery outcomes:
Clonotype Space: Projects the full repertoire view into an interactive landscape. Unlike static lists, it reveals distinct specificity clusters, allowing leads to be visually mapped against the global repertoire topology.
Algorithmic Lead Selection: Replaces traditional Top-N picking with a diversity-first algorithm. It prioritizes coverage of "Submerged" families, ensuring the final panel represents distinct, developable scaffolds rather than redundant dominant clones.
These components combine to form a standardized, high-throughput pipeline that converts raw FastQ files into a diversified, developable lead candidate table.
Building a Diversified Lead Panel
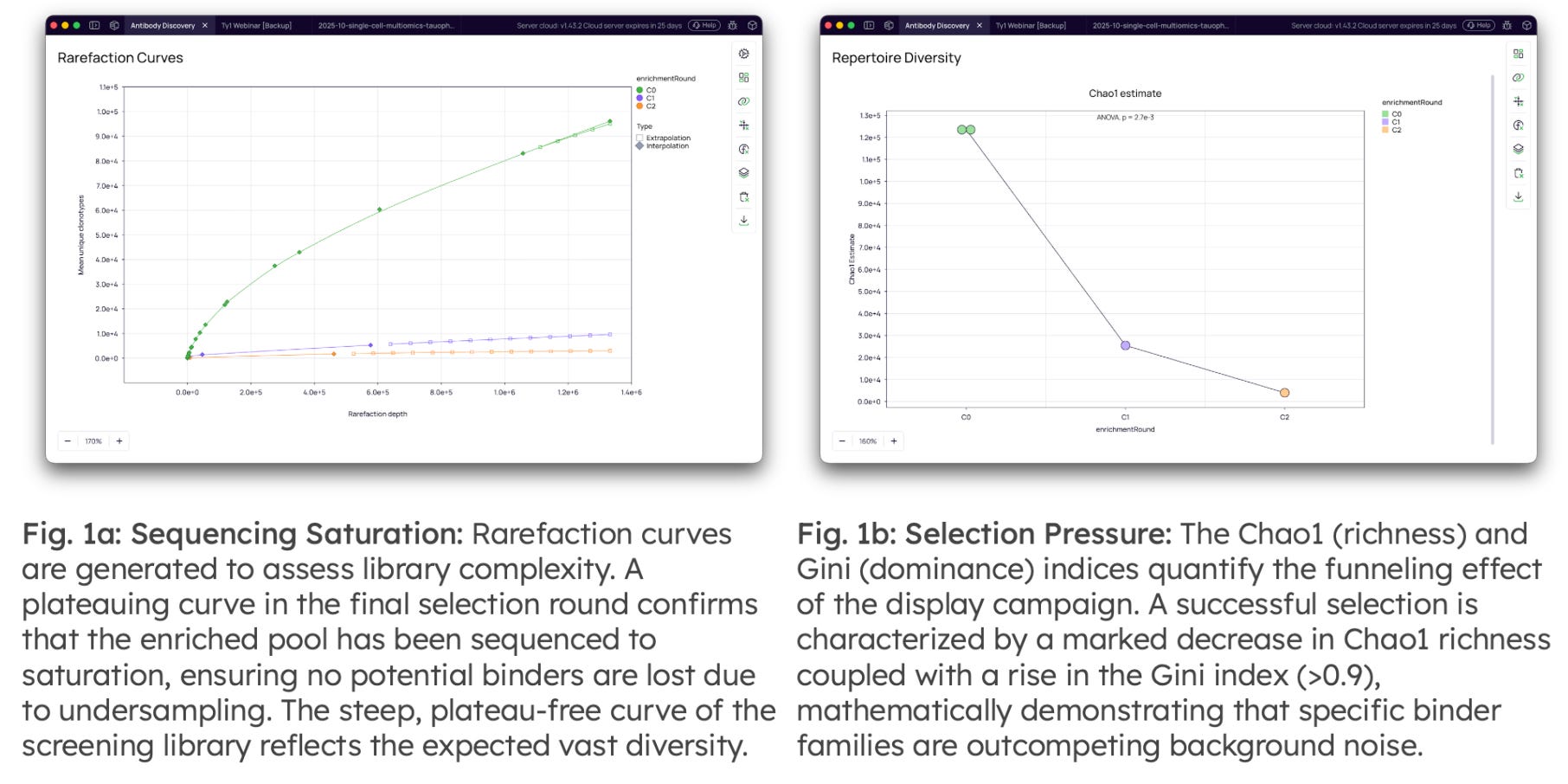
Validation of Sequencing Depth & Selection Pressure: Before clone/cluster-level analysis, the quality of the libraries and the efficacy of the selection pressure is validated statistically.
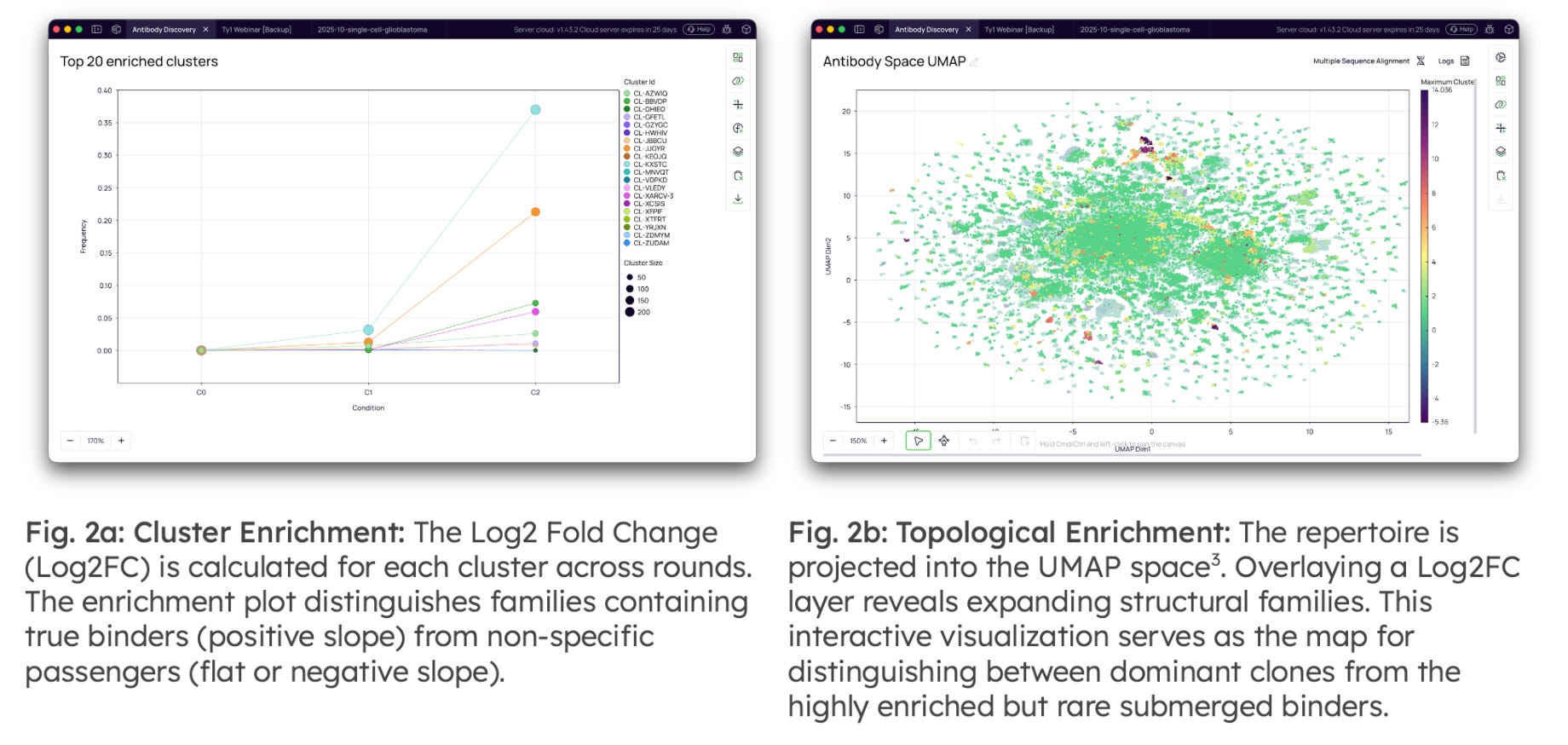
Antibody Clustering & Enrichment Analysis: Once data quality is established, the analysis focuses on identifying the "active" components of the repertoire. Functional clustering is performed by grouping clonotypes into families based on amino acid identity [2], reducing redundancy and allowing for the analysis of functional lineages rather than individual reads.
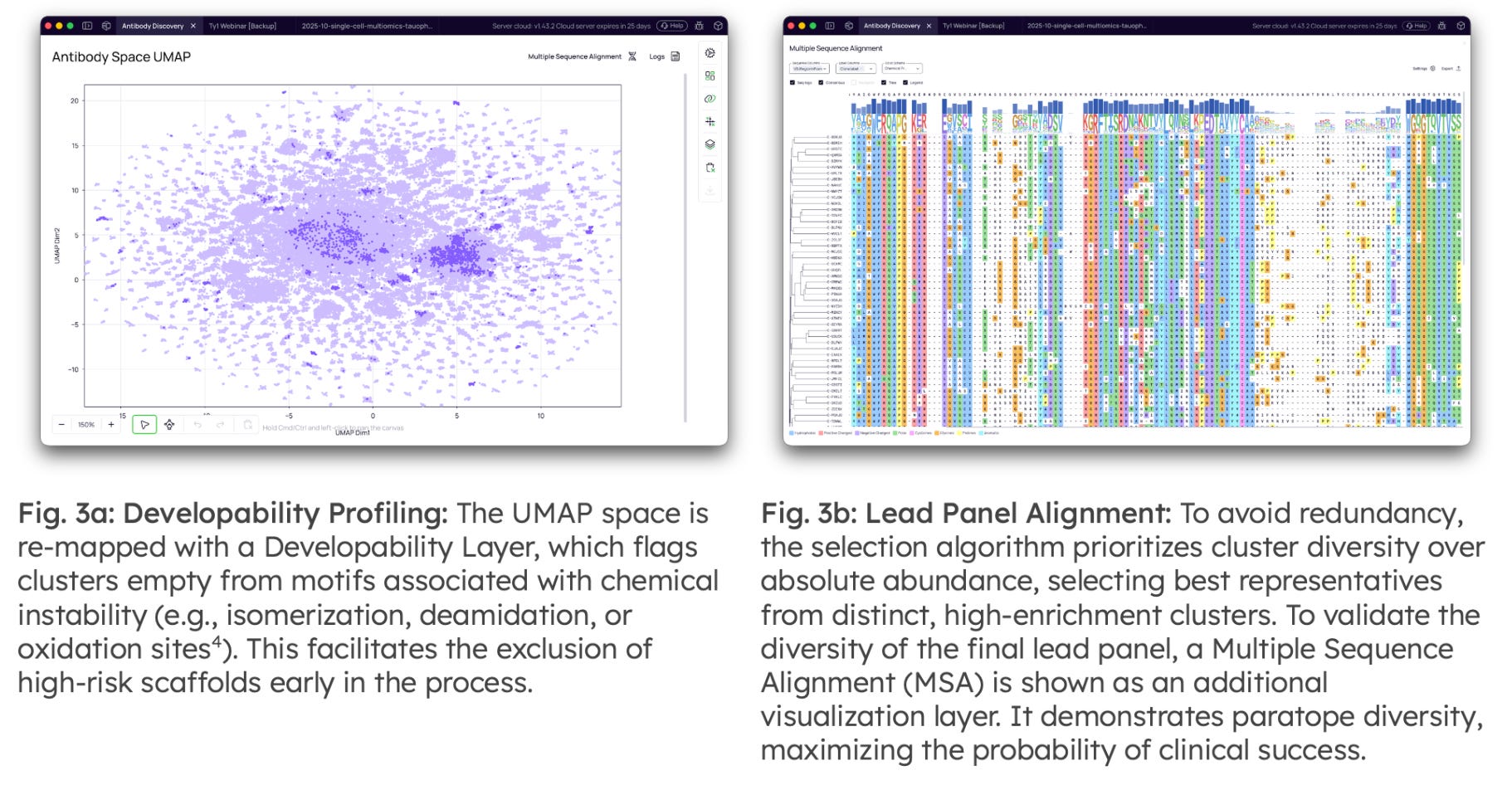
Developability Prediction & Lead Selection: The final stage refines the enriched pool into a diversified, low-risk panel for synthesis. To bridge the wet-lab/dry-lab divide, available screening data (e.g., ELISA or SPR hits from random colony picking) can be mapped directly to the NGS clusters. This validates the biological relevance of the computational families and allows for the discovery of superior somatic variants within the lineages of known binders.
Conclusion
Modern antibody discovery campaigns generate massive datasets that often outpace the analytical capabilities of fragmented toolchains. This Application Note introduces a unified, end-to-end computational workflow within Platforma designed to close the analysis gap between high-throughput Next-Generation Sequencing (NGS) and the selection of developable therapeutic leads.
By replacing isolated scripts and spreadsheets with a single, reproducible environment, this pipeline integrates rigorous upstream annotation, ultra-fast clustering and downstream functional validation. The workflow emphasizes algorithmic transparency and data provenance, allowing discovery teams to trace every candidate from its initial raw read to its final selection.
References:
[1] accessible at https://github.com/platforma-open/
[2] Clustering performed using MMSeqs2, Nat Biotechnol 35, 1026–1028 (2017). https://doi.org/10.1038/nbt.3988
[3] Journal of Open Source Software, 3(29), 861, https://doi.org/10.21105/joss.00861
[4] Proc. Natl. Acad. Sci. U.S.A. 114 (5) 944-949, https://doi.org/10.1073/pnas.1616408114