
Streamlining Peptide Discovery: From NGS to Functional Validation
High-Throughput In Vitro Peptide Discovery
Introduction
High-throughput peptide discovery aims to deliver a diversified panel of developable lead candidates. Traditional workflows often fail to meet this objective due to a critical analysis gap — the disconnect between massive NGS datasets and the limited capacity of downstream characterization. This Application Note outlines a scientifically validated workflow utilizing Platforma. By bridging upstream library profiling with downstream functional prediction, this protocol mitigates the risk of selecting redundant, non-specific, or liable peptides — ensuring the final candidate pool is maximized for both specificity and developability.
Necessity of NGS
Traditional peptide phage-display campaigns end with Sanger sequencing of a few hundred clones per round — vanishingly little against a 10⁸–10⁹ sequence library. After several rounds of panning, the picks converge onto one or two dominant peptides; the broader motif landscape stays invisible.
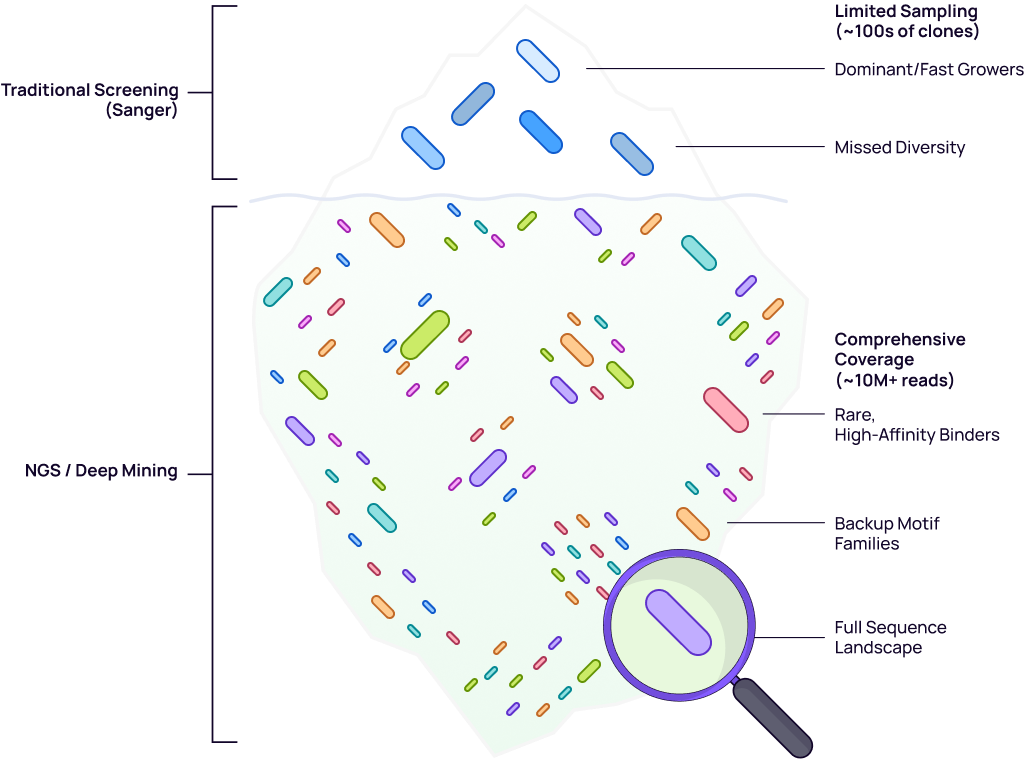
Deep NGS profiling (~10M+ reads per sample) replaces this with a quantitative picture. Every peptide’s frequency is measured across selection rounds, revealing a structured landscape of binding-motif families — each ranked by enrichment trajectory and filterable by sequence properties.
Methodology
This workflow enables the end-to-end peptide lead discovery in a managed computational environment. Prioritizing algorithmic transparency over black-box solutions, the system is built upon the Platforma SDK and the proprietary P-frame architecture (columnar, bioinformatics-native storage layer) to ensure data integrity and traceability.
The underlying logic is transparent and open source. At its core, the pipeline performs the sequence of steps for large-scale peptide discovery: profiling, clustering, enrichment analysis, and developability assessment. On top of this established workflow, Platforma introduces two innovations that significantly enhance discovery outcomes:
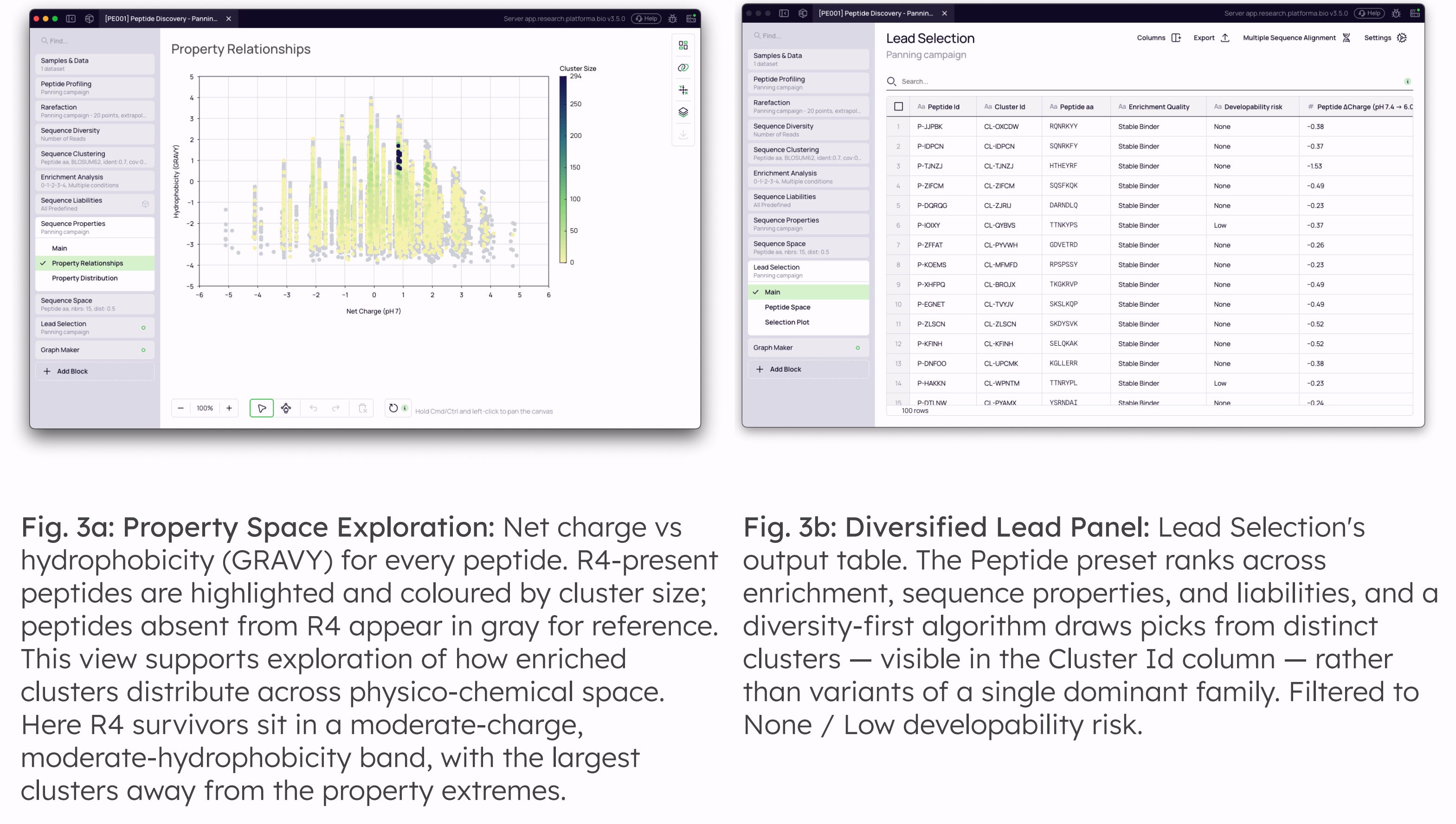
Sequence Properties: Net charge at pH 7, charge shift across the pH 7.4 → 6.0 window, isoelectric point, hydrophobicity (GRAVY), molecular weight, extinction coefficients, instability and aliphatic indices, aromaticity, and amino-acid composition are computed for every peptide and feed directly into Lead Selection as an integrated developability filter.
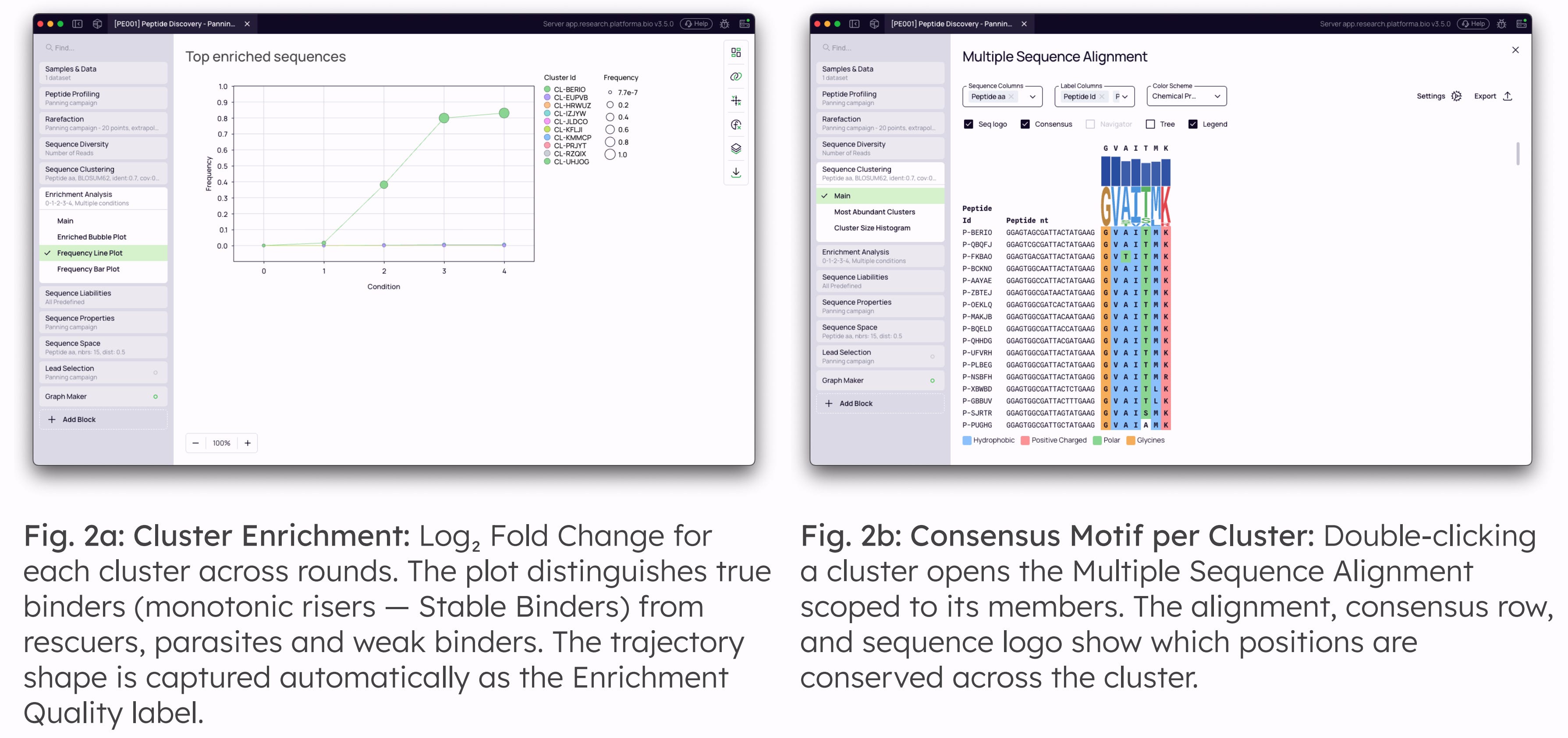
Enrichment Quality and Binding Specificity: Each peptide cluster is labeled by its trajectory shape across rounds (Stable Binder, Rescuer, Parasite, Weak Binder) and — when negative-control panning is included — by where its enrichment originates (Antigen-Specific, Non-Specific, Negative-Control, Not-Enriched) — ruling out fast-growing and surface-binding non-specifics at source.
These components combine to form a standardized, high-throughput pipeline that converts raw FASTQ files into a diversified, developable peptide lead candidate table.
Building a diversified lead pannel
Library Validation & Selection Pressure
Before cluster-level analysis, the quality of the libraries and the efficacy of the selection pressure are validated visually and statistically.
Peptide Clustering & Motif Discovery
Once library quality is established, peptides are grouped into clusters based on amino-acid sequence similarity using MMseqs2, reducing redundancy and enabling analysis at the level of consensus motifs rather than individual sequences. For very short peptide libraries (≤8 aa) we recommend selecting an exact-match similarity metric, preserving motif specificity at lengths where biochemical-similarity matrices become statistically noisy.
Developability Assessment & Lead Selection
The final stage refines the enriched library into a diversified, low-risk panel for synthesis. Enrichment-passing clusters are characterised quantitatively across physico-chemical and motif-based axes, and a diversity-first algorithm builds the picked panel from distinct motif families rather than collapsing onto the most enriched cluster alone.
Conclusion
Modern peptide discovery campaigns generate massive datasets that often outpace the analytical capabilities of fragmented toolchains. This Application Note introduces a unified, end-to-end computational workflow within Platforma designed to close the analysis gap between high-throughput Next-Generation Sequencing (NGS) and the selection of developable peptide leads.
By replacing isolated scripts and spreadsheets with a single, reproducible environment, this pipeline integrates rigorous upstream library profiling, ultra-fast clustering, multi-axis enrichment classification, and physico-chemical developability prediction. The workflow emphasizes algorithmic transparency and data provenance, allowing discovery teams to trace every candidate from its initial raw read to its final selection.
The protocol provides a standardized, scalable path to identifying a diversified panel of binding, developable peptide candidates that fully exploit the information content of the enriched library.